
M.A.S. Pinhel1, A.M. Crestani2, G.F. Sousa-Amorim3, M.L. Gregório4, J.C. Cação3, W.A. Tognola3, D. Rossi Silva Souza3
1. Ribeirao Preto Medical School – University of Sao Paulo, USP – Brazil; 2. Universidade Estadual Paulista (UNESP), Brazil; 3. Sao Jose do Rio Preto Medical School, FAMERP, SP, Brazil; 4. – Franca University – UNIFRAN, Franca, SP, Brazil
Corresponding Author: Marcela Augusta de Souza Pinhel, Faculdade de Medicina de Ribeirão Preto – USP, Avenida dos Bandeirantes, 3900, Ribeirao Preto – SP / CEP: 14090-900 / Brazil, Phone: +55 16 3315 4810, marcelapinhel@yahoo.com.br
J Aging Res Clin Practice 2017;inpress
Published online February 8, 2017, http://dx.doi.org/10.14283/jarcp.2017.3
Abstract
Background: Genetic polymorphisms in genes regulating cholesterol metabolism have been suggested to risk factor of developing Alzheimer’s disease (AD). Objective: to analyze the frequency of polymorphisms apolipoprotein E (APOE-HhaI) and adenosine triphosphate binding cassette transporter 1 (ABCA1-StyI) in patients with late-onset AD. Design: case-control study. Participants: We studied 166 subjects (≥65 years old): Study Group (SG)- 88 patients and Control Group (CG)- 88 without dementia. Setting: The polymorphisms were determined using the polymorphism chain reaction and restriction fragment length polymorphism (PCR-RFLP) methods. It was applied Fisher’s exact/chi-square tests (P<0.05). Results: Genotypes with APOE*4 prevailed in SG. The genotypic combination between APOE-HhaI and ABCA1-StyI polymorphisms showed a prevalence of heterozygous genotypes of risk for AD. Conclusion: Although genetic variants for ABCA1-StyI alone does not differentiate patients and controls, the G allele in synergy with APOE*4 allele is highlighted in patients suggesting the influence of ABCA1 in the disease.
Key words: Genetic variants, Alzheimer’s disease, ABCA1, APOE.
Background
Alzheimer’s disease (AD) is the most common form of dementia (1), affecting millions of individuals of all ethnic groups (2 to 6 million of North-Americans and about 1 million Brazilians), with higher incidence in Caucasian European descendants than in Japanese and black Africans (2). AD is characterized by deficits in short-term memory, language, visuospatial and executive functioning, eventually resulting in global cognitive impairment, with a mean disease duration of 5–15 years (3). Patients with AD present neuropathologic features like plaques formed by neurotoxic oligomeric aggregates of Ab42 peptide, produced by cleavage of the amyloid-beta precursor protein by β and γ secretases instead of by α secretase (3), and hyperphosphorylated forms of Tau protein aggregate into neurofibrillary tangles. The probable or possible diagnosis of AD is based on clinical criteria and may be supported by reduced amyloid-beta, increased total and hyperphosphorylated tau levels in the cerebrospinal fluid, hippocampal atrophy, reduced metabolism or retention of amyloid on image exams (4, 5).
Late-onset AD (starting from 65 years of age) corresponds to approximately 95% of the total cases, is well-defined by age factors as family history, Down syndrome and polymorphism of the apolipoprotein E (apo E) (2). In addition, there is crescent evidence that cholesterol plays an important role in the regulation of APP cleavage, held by the enzymes α and β secretase (6), besides the role of the APOE*4 allele in lipid metabolism (7).
These studies emphasize the critical involvement of cholesterol in the production of βA. However, there is few research about the mechanism by which cholesterol participates in this process. Studies suggest that the association between APP and platforms of lipids present in the plasma membrane determines the production of βA (6). These platforms are characterized as side groups of sphingolipids and cholesterol on the membrane (6). Together, these components build the platforms that are ordered and float in the lipid matrix of the cell membrane. Thus, APP is present in two cellular pathways, one associated with the platform of lipid in which the βA is generated, and other out of platforms, where the APP cleavage occurs by the β secretase enzyme (6). Therefore, it is possible that those platforms influence the production of βA.
Furthermore, genetic variants involved in the metabolism of lipoproteins and cholesterol, as well as the apoE, associated with receptors, proteins and enzymes have been highlighted as candidates for genetic risk factors for AD (8, 9). ApoE acts as a specific ligand for receptors such as the LDL receptor or apo B/E and LRP (LDL receptor-related protein), allowing the removal of particles, carrying them to the liver (10). APOE gene polymorphism was observed by Zannis et al. (11) in the form of three main alleles APOE*2, APOE*3 and APOE*4, that encode the isoforms of apo E designated by apoE2, apoE3 and apoE4. The APOE*4 allele is recognized for important role on development of AD, however the mechanism by which influences the disease is complex and obscure.
Several members of the ABC transporter superfamily form a complex of proteins that regulate homeostasis of cholesterol in cells and tissues using ATP to transport several molecules across biological membranes (7). The ABCs function in AD is relevant, especially for two prospects. First, these transporters participate in the efflux of excess cellular cholesterol, distributing these lipids to molecules that accept extracellular lipids, including apoE in the brain (7). We highlight the recognized impact of apoE in the deposition or removal of βA, and the important role of ABC transporters that mediate the transport of apoE affecting the pathogenesis of AD in vivo. Furthermore, ABC transporters also participate in the regulation of intracellular cholesterol levels. Several studies support this hypothesis, using cell models, indicating that high intracellular concentration of cholesterol increases the amyloidogenic process of APP raising the production of βA (7, 12). Moreover, depletion of intracellular cholesterol is associated with reduced production of βA and tendency for the non-amyloidogenic process of APP (6).
ABCA1 (adenosine triphosphate binding cassette transporter 1) has the primary function of cellular cholesterol efflux and apolipoproteins, including apoA-I, which acts in the biosynthesis of HDL in the peripheral circulation, besides the secretion of apoE in astrocytes and microglia (15). In this context, ABCA1 is expressed in brain and apoE is the largest lipid acceptor that connects the efflux of cholesterol via ABCA1 in the CNS (7, 16, 17). The gene encoding ABCA1 is located on human chromosome 9q22 (18). A polymorphism for ABCA1, also known as R219K, promotes a nucleotide substitution between G>A in exon 7, resulting in the change from lysine to arginine at 219 position, a region in which occurs the first extracellular loop of ABCA1 protein.
Therefore, this study aimed to analyze the allelic and genotypic distribution of APOE-Hha I and ABCA1-Sty I polymorphisms in patients with late-onset AD; evaluating together the alleles, which represents risk, intending to identify a subgroup at risk of AD.
Methods
We studied 166 individuals of mixed race (19), over age 65, regardless of gender, divided into two groups, including Study Group (SG) – 88 patients with late-onset AD (76.6 ± 11.8 years and 68% female) and Control Group (CG) – 88 elderly patients without dementia (72.3 ± 6.76 years, 61% female). Patients were treated at the Geriatric Neurological Clinic of the Hospital Base of the Sao Jose do Rio Preto Medical School HB/FAMERP. The diagnosis of AD followed the criteria of service of Neurogeriatrics (NINCDS – ADRDA) (4, 5), including neuropsychological test organized according to the protocol used in that service and at least one method of neuroimaging (computed tomography, magnetic resonance imaging cerebral or single photon emission) consistent with the diagnosis. Individuals with other possible etiologies for dementia were excluded. The control group, including elderly people with the same age in group of patients, without cognitive impairment, was from support groups, held in that institution. This study was established as part of a project evaluated and approved by Certificate of Appreciation Presentation Ethics (CEP-FAMERP – CAAE: 0046.0.140.000-08). All subjects signed an informed consent form. For genotyping, DNA was extracted from peripheral whole blood using a saline precipitation method or saling-out (20).
The APOE-HhaI (rs429358 and rs7412), and ABCA1-StyI (rs2230806) polymorphisms were determined using the polymorphism chain reaction and restriction fragment length polymorphism (PCR-RFLP) methods (Mastercycler – Eppendorf) with restriction enzymes HhaI and StyI, respectively (Fermentas). The PCR solution was comprised of 2.5 μL of desoxynucleotide (4mM), 2.5uL of dimethylsulfoxide 10%, 2.5 uL of each primer (25mM), 0,2 uL of Taq polymerase (5U/uL), 11 uL of Milli Q water an 2.5 uL of diluted genomic DNA (0.25 ug).
For APOE-HhaI polymorphism was amplified region of the 112 and 158 polymorphic codons. In this case, we used the primers P1:5′ ACAGAATTCGCCGGCCTGGTACAC3′ and P2:5′ TAAGCTTGGCACGGCTGTCCAGCA3′. The initial denaturation of DNA was obtained at 94°C for 5 minutes and the reaction mixture was then subjected to 40 cycles of 94°C for 30 seconds and 65°C for 2 minutes with the final cycle at 72°C for 7 minutes (21). The product of PCR amplification was subjected to HhaI restriction enzyme (5U per reaction tube) in water bath at 37 ° C, overnight, to divide the amplified sequences of APOE*2, APOE*3 and APOE*4 alleles in specific regions (GCGC), separating fragments with 91pb and 83bp (APOE*2), 91bp and 48pb (APOE*3) and 72bp and 48bp (APOE*4). Electrophoresis was performed in 6% polyacrylamide non-denatured gel under constant current of 200V for 1 hour and 30 minutes. As a control we used a sample of standard 100bp DNA (Fermentas) (21).
To the ABCA1-Sty I polymorphism, we used these primers in PCR: P1: 5’CCTGTCATTGTGCCTTGT-3 ‘and P2: 5′-GGATTGGCTTCAGGATGT-3’. The initial denaturation was performed at 98°C for 3 minutes, followed by 32 cycles of denaturation at 94°C for 45 seconds, annealing at 58 ° C for 1 minute and 30 seconds and extension at 72°C for 1 minute and 30 seconds, with the final cycle at 72°C for 10 minutes. The amplification product was subjected to enzymatic restriction for 4 hours at 37°C with StyI endonuclease. The digested DNA samples and a sample of standard DNA (100bp ladder – Fermentas Life Sciences) were applied to agarose gel 1% and subjected to electrophoresis to identify the polymorphisms. For ABCA1-Sty I polymorphism is possible to identify fragments with 183 base pairs (bp) (A allele) and G allele with fragments of 131 and 112 bp (22).
Biochemcial evaluation consisted of the determination of serum concentrations of total cholesterol (TC), low density lipoprotein cholesterol (LDLc), high density lipoprotein cholesterol (HDLc) and triglycerides (TG) using the reference values of the I Brazilian Directives for Cardiovascular Disease Prevention (23).
Statistical Methodology
Fisher’s exact test and chi-square (x²) were applied in the analysis of allelic and genotypic distributions of APOE-HhaI and ABCA1-StyI, as well as in calculations of Hardy-Weinberg equilibrium, considering the distribution of genotypes for these polymorphisms. The t test was applied in the analysis of lipidic profile. It was admitted an alpha error of 5%. Statistical Package for Social Science 20.0 software (SPSS [Inc. Chicago. IL]).
Results
The APOE*3/3 genotype prevailed in GC (83%) compared to SG (62%, P = 0.004), while the mutant heterozygous model (APOE*_/4) stands out on patients (36% versus CG = 8.5%, P = 0.0001). Thus, the APOE*3 allele showed a higher frequency in controls (0.90 versus SG = 0.80, P = 0.011), while APOE*4 prevailed in SG (0.19 vs. CG = 0.055, P = 0.0003) (Table 1).
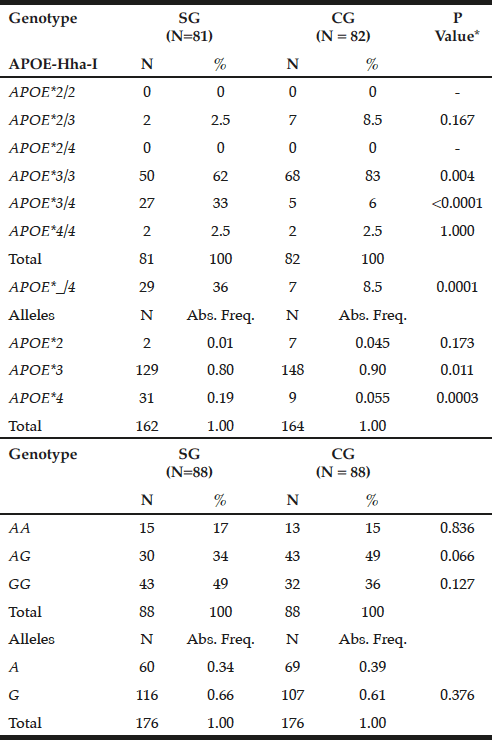
Table 1
Genotypic and allelic frequency distribution for APOE-Hha-I and ABCA1-Sty I polymorphisms in patients with late-onset Alzheimer’s disease (SG) and individuals without clinical signs of the disease (CG)
*Chi-square or Fisher test; N= number of individuals; Abs. Freq.= absolute frequency; ABCA1= adenosine triphosphate binding cassette transporter 1; ApoE= apolipoprotein E; P= Level of significance <0.05; SG= study group; CG= control group.
For ABCA1-StyI polymorphism there was similarity in allelic and genotypic distribution between the groups (P> 0.05), highlighting the GG genotype in patients (49%) and AG in controls (49%), with prevalence of the G allele in both groups (0.66 and 0.61, respectively, p = 0.376, Table 1).
Table 2 shows the genotypic combination between APOE-HhaI polymorphism and ABCA1-StyI. The APOE*_/4 + GG genotypes prevailed in patients (20% versus GC = 5%, P = 0.0041), as observed for APOE*_/4 + _/G (SG = 28% versus GC = 8.5%, P = 0.0021).
It was found Hardy-Weinberg equilibrium for APOE-HhaI in patients (x²= 0.55; P= 0.50), which did not occur in controls (x2= 39.35; P <0.0001). For ABCA1-Sty I the Hardy-Weinberg equilibrium was maintained in controls (x2= 0.06; P = 0.90), but not in patients (x2= 5.13; P= 0.025).
The lipid profile was similar between the groups with emphasis on increased levels of TC and LDLc in patients (207.3±47.8 mg/dL and 120.3±46.5 mg/dL, respectively) compared with the control group (187.2±60.2 mg/dL and 102.9±51.9mg/dL P=0.014, P=0.022, respectively). However, with the exception of TC in patients, lipid profile values remained within normal limits (Table 3) (23).
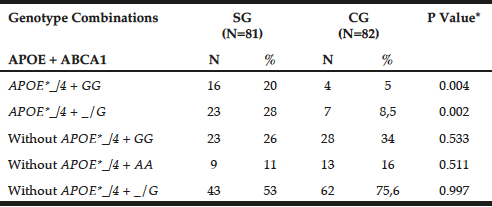
Table 2
Preferential combination between APOE-Hha I and ABCA1-Sty I polymorphisms in patients with late-onset Alzheimer’s disease (SG) and individuals without clinical signs of the disease (CG)
*Chi-square or Fisher test; N= number of individuals; Abs. Freq.= absolute frequency; ABCA1= adenosine triphosphate binding cassette transporter 1; ApoE= apolipoprotein E; P= Level of significance <0.05; SG= study group; CG= control group.
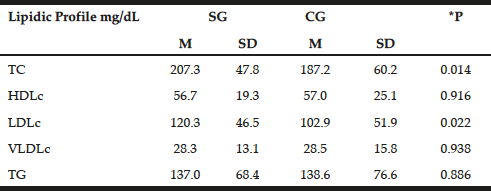
Table 3
Distribution of mean values, standard deviations and mean differences for biochemist profile in patients with late-onset Alzheimer disease (SG) and individuals without clinical signs of disease (Control – CG)
t test; *P = significance level P <0.05; TC = total cholesterol, HDLc = concentration of high density lipoprotein cholesterol; LDLc = concentration of low-density lipoprotein cholesterol; VLDLc = concentration of very low density lipoprotein cholesterol; TG = triglycerides; M = Average; SD = standard deviation; SE = study group; CG = control group.
Discussion
The present study confirms the association of APOE-HhaI polymorphism with late onset AD, and suggests synergism of APOE*4 allele and G to ABCA1 in the metabolism of lipids in individuals with AD. In this case, the APOE-HhaI allelic and genotypic distribution discriminates patients with high frequency of APOE*4 allele (0.19) and controls (0.055, P= 0.0007). These results are consistent with other studies in Brazilian papers, whose frequency of the referred allele ranged from 0.22 to 0.34, whereas, in control group, the values remained between 0.07 and 0.11 (2, 24), and also consistent with results of studies with patient populations in Colombia, where the values for APOE*4 allele frequency was between 0.23 and 0.08 for study and control group, respectively (25).
The ABCA1-StyI polymorphism, however, was not associated with late onset AD. The G allele frequency was high in this casuistic study with AD (0.66), differing from the world’s population, including Chinese casuistry (0.48) (26), American and Canadian (0.27 for both) (27).
A study by Wavrant-De Vrieze et al. (28) in Spanish casuistic sample with AD, showed association between R219K polymorphism (ABCA1-StyI) and AD (P = 0.010), regardless of the presence of the APOE*4 allele. In addition, a sample of 1275 Swedish individuals with AD and control 2203 also discloses the influence of R219K polymorphism (AB 1.25, CI 1.12 to 1.40, P = 5.9 × 10 5), which is linked to G allele, associated with increased risk for AD (29). However, there is disagreement among authors.
Such polymorphism has been extensively studied in relation to risk for AD among other phenotypes, in particular those related to cardiovascular disease and plasma lipids (29). There is reference to the G allele for ABCA1 may amount to 2.77 times the risk for AD (12) and this variant can influence levels of 24S-hydroxycholesterol, which is essential for the transfer of cholesterol in blood-brain barrier increasing, thereby, the βA42 producing (30).
The present study shows an association between the synergism of APOE*_/4 and _/G genotypes and late onset AD, corroborating with the study conducted for the first time in Hispanic casuistic sample, showing that carriers of the APOE*4 allele and G allele for ABCA1 are at risk for increased AD by 3.7 times (15). In this case, the G allele represents the reduced activity of gene expression and thus could modify the risk for AD in synergism with allele APOE*4 (28, 29). On the other hand, Wang & Jia (26) revealed that the GG genotype for ABCA1 may reduce the risk for AD in Chinese sample. However, this association was only observed in women (P= 0.016) or individuals without the APOE*4 allele (P= 0.010).
In this context, there is reference of ABCA1 as a regulator of lipidation and apoE levels in brain. Deficiency of ABCA1 leads to loss of approximately 80% of apoE in the brain and the rest of residual apoE is poorly lipidated (17). In this case, studies in animal models of AD have shown that apoE poorly lipidated increases the amyloid load. This reinforces the hypothesis that apoE lipidation by ABCA1 affects deposition or removal of the amyloid protein, contributing to the formation of senile plaques in excess (17). Conversely, overexpression of the brain ABCA1 promotes apoE lipidation and eliminates the formation of mature amyloid plaques (17). These studies show that the capacity lipidic binding of apoE is the primary mechanism to the pathogenesis of AD.
This study highlights the demonstration of synergism between APOE and ABCA1, represented by the APOE*4 and G allele, respectively, with possible impact on the risk for late onset AD. The reduced expression of ABCA1 due to the presence of the G allele, may be compounded by low cholesterol efflux of glial cells to neurons due to the presence of the APOE*4 allele, which promotes the outflow less efficient compared to the APOE*2 and APOE*3 alleles (17).
Among patients with AD was found that the genotype distribution for APOE-HhaI was in agreement with Hardy-Weinberg equilibrium, the same was not true for the control group, as compared to ABCA1-Sty I polymorphism in patients, as was also observed in some studies of case-control analyzes of different genetic polymorphisms (29). The criterion for selecting the groups used in this study provided a group of older individuals, given that late-onset AD occurs in individuals of older age. Thus, the profile of patients, as well as the control group does not represent the general population by gender and age, which could influence the distribution of genotypes between the groups. Incidentally, the absence of Hardy-Weinberg equilibrium would be expected for a wide range of genetic diseases, considering the contribution of genes, though modest, for complex diseases. However, considering the large number of candidate gene studies in different cases, the number of genetic markers in imbalance is scarce. In this case, it is possible that researchers disregard the distribution of imbalanced genotypes, neglecting valuable information to identify casual polymorphisms (30).
Conclusion
The APOE-HhaI polymorphism is associated with late onset AD, however, genetic variants in ABCA1-StyI alone do not differentiate patients and controls. Furthermore, APOE*4 and G alleles in synergism are related to AD. Though, studies with more numerous series are needed to confirm this combination of genes with AD, as well as its influence on lipid metabolism.
Funding: This work was supported by The Sao Paulo Research Foundation (FAPESP) (#grant 2010/17476-0) and São Jose do Rio Preto Medical School (FAMERP – Laboratory of Biochemistry and Molecular Biology).
Acknowledgements: nothing to declare.
Ethical standard: This project was evaluated and approved by Certificate of Appreciation Presentation Ethics (CEP-FAMERP – CAAE: 0046.0.140.000-08). This is according to DECLARATION OF HELSINKI. Ethical Principles for Medical Research Involving Human Subjects
References
1. Lupton MK, Proitsi P, Lin K, et al. The role of ABCA1 gene sequence variants on risk of Alzheimer’s disease. J Alzheimers Dis 2014;38:897-906.
2. Pinhel MA, Nakazone MA, Cação JC, et al. Glutathione S-transferase variants increase susceptibility for late-onset Alzheimer’s disease: association study and relationship with apolipoprotein E epsilon4 allele. Clin Chem Lab Med 2008;46:439-445.
3. Cohn-Hokke PE, Elting MW, Pijnenburg YA, et al. Genetics of dementia: update and guidelines for the clinician. Am J Med Genet B Neuropsychiatr Genet 2012; 159B:628-643.
4. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer Dement 2011; 7:263-269.
5. McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS–ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34:939–944.
6. Ehehalt R, Keller P, Haass C, et al. Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipids rafts. J Cell Biol 2003;160:113-123.
7. Hirsch-Reinshagen V, Burgess LB, Wellington CL. Why lipids are important for Alzheimer disease? Mol Cell Biochem 2009;326:121–129.
8. Lupton MK, Proitsi P, Lin K, et al. The role of ABCA1 gene sequence variants on risk of Alzheimer’s disease. J Alzheimers Dis 2014;38:897-906.
9. Xue-Shan Z, Juan P, Qi W, et al. Imbalanced cholesterol metabolism in Alzheimer’s disease. Clin Chim Acta 2016;456:107-114.
10. Harris JR, Milton NG. Cholesterol in Alzheimer’s disease and other amyloidogenic disorders. Subcell Biochem 2010;51:47-75.
11. Zannis VI, Just PW, Breslow JL. Human apolipoprotein E isoprotein subclasses are genetically determined. Am J Hum Genet 1981;33:11-24.
12. Rodríguez-Rodríguez E, Mateo I, Infante J, et al. Interaction between HMGCR and ABCA1 cholesterol-related genes modulates Alzheimer’s disease risk. Brain Res 2009;1280:166-171.
13. Refolo LM, Malester B, LaFrancois J, et al. Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol Dis 2000;7:321-331.
14. Hirsch-Reinshagen V, Wellington CL. Cholesterol metabolism, apolipoprotein E, adenosine triphosphate-binding cassette transporters, and Alzheimer’s disease. Curr Opin Lipidol 2007;18:325-332.
15. Wahrle SE, Jiang H, Parsadanian M, et al. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem 2004;279:40987-40993.
16. Burns MP, Vardanian L, Pajoohesh-Gangi A. The effects of ABCA1 on cholesterol efflux and Ab levels in vitro and in vivo. J Neurochem 2006;98:792–800.
17. Blacker D, Bertram L, Saunders AJ, et al. Results of a high-resolution genome screen of 437 Alzheimer disease families. Hum Mol Genet 2007;12:23-32.
18. Pena SD, Bastos-Rodrigues L, Pimenta JR, et al. DNA tests probe the genomic anscestry of Brazilians. Braz J Med Biol Res 2009;42:870-876.
19. Salazar LA, Hirata MH, Cavalli SA, et al. Optimized procedure for DNA isolation from fresh and cryopreserved clotted human blood useful in clinical molecular testing. Clin Chem 1998;44:1748-1750.
20. Hixson JP, Vernier DT. Restriction isotyping of human apoliprotein E by gene amplification and cleavage with Hhal. J Lipid Res 1990;31:545-548.
21. Fidani L, Goulas A, Crook R, et al. An association study cholesteryl ester transfer protein TaqI B polymorphism with late onset Alzheimer’s disease. Neurosci Lett 2004;357:152-154.
22. Simão AF, Precoma DB, Andrade JP, et al. I Diretriz Brasileira de Prevenção Cardiovascular. Arq Bras Cardiol 2013;101(6 Suppl 2):1-63.
23. Souza DRS, Godoy MRP, Hotta J, et al. Association of apoliprotein E polymorfism in late-onset Alzheimer’s disease and vascular dementia in Brazilians. Bras J Med Biol Res 2003;36:1-5.
24. Jacquier M, Arango D, Villareal E, et al. Apo e4 and Alzheimer’s disease. Positive association in a Colombian clinical series and review of the Latin-American studies. Arq Neuropsiquiatr 2001;59:11-17.
25. Wang F, Jia J. Polymorphisms of cholesterol metabolism genes CYP46 and ABCA1 and the risk of sporadic Alzheimer’s disease in Chinese. Brain Res 2007;1147:34-38.
26. Li Y, Tacey K, Doil L, et al. Association of ABCA1 with late-onset Alzheimer’s disease is not observed in a case–control study. Neurosci Lett 2004;366:268–271.
27. Wavrant-De Vrièze F, Compton D, Womick M, et al. ABCA1 polymorphisms and Alzheimer’s disease. Neurosci Lett 2007;416:180-183.
28. Reynolds CA, Hong MG, Eriksson UK, et al. A survey of ABCA1 sequence variation confirms association with dementia. Hum Mutat 2009;30:1348-1354.
29. Kölsch H, Lüjohann D, Jessen F, et al. Polymorphism in ABCA1 influences CSF 24Shydroxycholesterol levels but is not a major risk factor of Alzheimer’s disease. Int J Mol Med 2006;17:791–794.
30. Xu J, Turner A, Little J, et al. Positive results in association studies are associated with departure from Hardy-Weinberg equilibrium: hint for genotyping error? Hum Genet 2002;111:573-574.